
Όλα τα περιεχόμενα του iLive ελέγχονται ιατρικά ή ελέγχονται για να διασφαλιστεί η όσο το δυνατόν ακριβέστερη ακρίβεια.
Έχουμε αυστηρές κατευθυντήριες γραμμές προμήθειας και συνδέουμε μόνο με αξιόπιστους δικτυακούς τόπους πολυμέσων, ακαδημαϊκά ερευνητικά ιδρύματα και, όπου είναι δυνατόν, ιατρικά επισκοπικά μελέτες. Σημειώστε ότι οι αριθμοί στις παρενθέσεις ([1], [2], κλπ.) Είναι σύνδεσμοι με τις οποίες μπορείτε να κάνετε κλικ σε αυτές τις μελέτες.
Εάν πιστεύετε ότι κάποιο από το περιεχόμενό μας είναι ανακριβές, παρωχημένο ή αμφισβητήσιμο, παρακαλώ επιλέξτε το και πατήστε Ctrl + Enter.
Νόσος του Huntington
Ιατρικός εμπειρογνώμονας του άρθρου

Τελευταία επισκόπηση: 05.07.2025
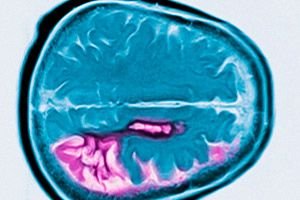
Η νόσος του Χάντινγκτον είναι μια αυτοσωμική επικρατής νευροεκφυλιστική διαταραχή που χαρακτηρίζεται από προοδευτική γνωστική έκπτωση, ακούσιες κινήσεις και μειωμένο κινητικό συντονισμό που ξεκινά στη μέση ηλικία. Η διάγνωση επιβεβαιώνεται με γενετικό έλεγχο. Η θεραπεία είναι κυρίως συμπτωματική. Ο γενετικός έλεγχος μπορεί να συνιστάται για συγγενείς εξ αίματος. Ο Τζορτζ Χάντινγκτον περιέγραψε για πρώτη φορά την πάθηση το 1872, αφού μελέτησε μια οικογενειακή περίπτωση κατοίκων του Λονγκ Άιλαντ.
Η συχνότητα εμφάνισης της νόσου του Huntington είναι περίπου 10 περιπτώσεις ανά 100.000 κατοίκους και, δεδομένης της όψιμης εμφάνισής της, περίπου 30 άτομα στα 100.000 έχουν 50% κίνδυνο να την εμφανίσουν στη ζωή τους. Αν και η νόσος εμφανίζεται συχνότερα μεταξύ των ηλικιών 35 και 40 ετών, το ηλικιακό εύρος εμφάνισης είναι αρκετά ευρύ, με την πρωιμότερη εμφάνιση να είναι στην ηλικία των 3 ετών και την τελευταία στην ηλικία των 90 ετών. Αν και αρχικά θεωρούνταν ότι η νόσος είχε 100% διεισδυτικότητα, τώρα πιστεύεται ότι αυτό δεν ισχύει πάντα. Σε άτομα που κληρονόμησαν το γονίδιο για τη νόσο από τον πατέρα τους, η νόσος εκδηλώνεται κατά μέσο όρο 3 χρόνια νωρίτερα από ό,τι σε εκείνα που κληρονόμησαν το παθολογικό γονίδιο από τη μητέρα τους. Σε περίπου 80% των ασθενών που κληρονόμησαν το παθολογικό γονίδιο από τον πατέρα τους, η νόσος εκδηλώνεται πριν από την ηλικία των 20 ετών. Το φαινόμενο της πρωιμότερης εκδήλωσης ενός γενετικού ελαττώματος στους απογόνους ονομάζεται προσμονή.
 [ 1 ]
[ 1 ]
Τι προκαλεί τη νόσο του Χάντινγκτον;
Η νόσος του Huntington δεν έχει προτίμηση φύλου. Εμφανίζεται ατροφία του κερκοφόρου πυρήνα, όπου μικροί νευρώνες εκφυλίζονται και το επίπεδο των νευροδιαβιβαστών - γ-αμινοβουτυρικό οξύ (GABA) και ουσία P - μειώνεται.
Ένα μεταλλαγμένο γονίδιο με αυξημένο αριθμό («επέκταση») αλληλουχιών DNA CAG (κυστεΐνη-αλανίνη-γλυκίνη) που κωδικοποιούν το αμινοξύ γλουταμίνη είναι υπεύθυνο για την ανάπτυξη της νόσου του Huntington. Το προϊόν αυτού του γονιδίου, η μεγάλη πρωτεΐνη huntingtin, περιέχει υπερβολική ποσότητα υπολειμμάτων πολυγλουταμίνης, η οποία οδηγεί στην ασθένεια με άγνωστο μηχανισμό. Όσο περισσότερες επαναλήψεις CAG, τόσο νωρίτερα εμφανίζεται η ασθένεια και τόσο πιο σοβαρή είναι η εξέλιξή της. Από γενιά σε γενιά, ο αριθμός των επαναλήψεων μπορεί να αυξηθεί, γεγονός που με την πάροδο του χρόνου οδηγεί σε επιδείνωση του οικογενειακού φαινοτύπου.
Παρά το σημαντικό ενδιαφέρον για τις γενετικές και βιοχημικές αλλαγές στη νόσο του Πάρκινσον, η αναζήτηση ενός γονιδίου για τη νόσο ήταν ανεπιτυχής μέχρι τα τέλη της δεκαετίας του 1970. Εκείνη την εποχή, η Nancy Wexler και ο Allan Tobin οργάνωσαν ένα εργαστήριο που χρηματοδοτήθηκε από το Ίδρυμα Κληρονομικών Νοσημάτων για να συζητήσουν μια στρατηγική για την εύρεση ενός γονιδίου για τη νόσο του Huntington. Οι David Housman, David Botstein και Ray White, που παρευρέθηκαν στη συνάντηση, πρότειναν ότι οι πρόσφατα αναπτυγμένες τεχνικές ανασυνδυασμένου DNA θα μπορούσαν να βοηθήσουν στην επίτευξη αυτού του στόχου. Ένα βασικό έργο του έργου ήταν η εύρεση μιας μεγάλης οικογένειας με πολλές γενιές της νόσου του Huntington για τη λήψη δειγμάτων DNA. Το 1979, ξεκίνησε ένα κοινό έργο επιστημόνων από τη Βενεζουέλα και τις Ηνωμένες Πολιτείες για να εξετάσουν μια μεγάλη οικογένεια με νόσο του Huntington που ζούσε στις όχθες της λίμνης Maracheibo (Βενεζουέλα). Το 1983, το γονίδιο της νόσου του Huntington εντοπίστηκε στο τέλος του βραχέος βραχίονα του χρωμοσώματος 4 (Gusella et al., 1983) και μια δεκαετία αργότερα αποκαλύφθηκε ότι η μετάλλαξη αυτού του γονιδίου συνίσταται σε αύξηση του αριθμού των επαναλήψεων του τρινουκλεοτιδίου κυτοσίνης-αδενίνης-γουανίνης (CAG) (Huntington's Disease Collaborative Research Group, 1993). Η μεθοδολογία που αναπτύχθηκε από αυτήν την επιστημονική ομάδα θεωρείται επί του παρόντος πρότυπο για την κλωνοποίηση θέσης νέων γονιδίων.
Ενώ το γονίδιο άγριου τύπου έχει ένα εύρος 10-28 επαναλήψεων CAG, η μεταλλαγμένη μορφή του γονιδίου που προκαλεί τη νόσο του Huntington έχει ένα αυξημένο εύρος από 39 σε περισσότερες από 100 επαναλήψεις CAG. Η ανακάλυψη της επέκτασης των τρινουκλεοτιδικών επαναλήψεων έχει βοηθήσει στην εξήγηση πολλών από τα κλινικά χαρακτηριστικά της νόσου. Συγκεκριμένα, διαπιστώθηκε μια αντίστροφη συσχέτιση μεταξύ της ηλικίας έναρξης και του μήκους της περιοχής με επαναλαμβανόμενα τρινουκλεοτίδια. Η πρόβλεψη της πατρικής κληρονομικότητας μπορεί να εξηγηθεί από το γεγονός ότι η αύξηση στον αριθμό των επαναλήψεων εμφανίζεται συχνά στους άνδρες κατά τη σπερματογένεση. Η ανάλυση νέων μεταλλάξεων έχει δείξει ότι συνήθως εμφανίζονται όταν ένας από τους γονείς, συνήθως ο πατέρας, είχε αριθμό επαναλήψεων CAG υψηλότερο από 28. Σε αυτήν την περίπτωση, ο αριθμός αυτών των επαναλήψεων αυξήθηκε στην επόμενη γενιά. Έχει πλέον διαπιστωθεί ότι εάν ο αριθμός των επαναλήψεων δεν είναι μεγαλύτερος από 28, μεταδίδεται σταθερά από γενιά σε γενιά. Εάν ο αριθμός των επαναλήψεων είναι από 29 έως 35, τότε τα συμπτώματα της νόσου του Huntington δεν εμφανίζονται, αλλά όταν μεταδίδεται στους απογόνους, το μήκος αυτής της περιοχής μπορεί να αυξηθεί. Εάν ο αριθμός των επαναλήψεων είναι από 36 έως 39, τότε σε ορισμένες περιπτώσεις (αλλά όχι πάντα) η ασθένεια μπορεί να εκδηλωθεί κλινικά (ατελής διείσδυση) και όταν μεταδίδεται στους απογόνους, είναι πιθανή η αύξηση του αριθμού των τρινουκλεοτιδικών επαναλήψεων. Εάν ο αριθμός των επαναλήψεων υπερβαίνει τις 40, τότε η ασθένεια εμφανίζεται σε όλες σχεδόν τις περιπτώσεις και όταν μεταδίδεται στους απογόνους, είναι πιθανή η περαιτέρω επέκταση των επαναλήψεων. Οι λόγοι για την αύξηση του αριθμού των επαναλήψεων παραμένουν άγνωστοι.
Παθομορφολογία της νόσου του Huntington
Η νόσος του Huntington χαρακτηρίζεται από νευρωνική απώλεια κυρίως στον κερκοφόρο πυρήνα και το κέλυφος, και σε κάποιο βαθμό και στον φλοιό και σε άλλες δομές του εγκεφάλου. Το συνολικό βάρος του εγκεφάλου στη νόσο του Huntington μειώνεται όχι μόνο από τη μείωση του αριθμού των νευρώνων, αλλά και από την απώλεια λευκής ουσίας. Στον εγκεφαλικό φλοιό, τα κύτταρα στις στιβάδες V και VI επηρεάζονται περισσότερο. Η σοβαρότητα των μικρο- και μακροσκοπικών εκφυλιστικών αλλαγών (προσαρμοσμένων για την ηλικία θανάτου) συσχετίζεται με τον αριθμό των επαναλήψεων CAG. Λεπτομερής παθολογοανατομική ανάλυση αλλαγών σε αρκετές εκατοντάδες περιπτώσεις νόσου του Huntington έχει δείξει ότι η εκφύλιση του ραβδωτού σώματος ξεκινά στο ραχιαίο-μεσαίο τμήμα του κερκοφόρου πυρήνα και στο ραχιαίο-πλάγιο τμήμα του κέλυφους και στη συνέχεια εξαπλώνεται κοιλιακά. Διαφορετικές ομάδες νευρώνων στον κερκοφόρο πυρήνα και το κέλυφος επηρεάζονται σε διαφορετικό βαθμό. Οι ενδιάμεσοι νευρώνες στο ραβδωτό σώμα παραμένουν σχετικά άθικτοι, αλλά ορισμένοι νευρώνες προβολής επηρεάζονται επιλεκτικά. Στη νεανική μορφή της νόσου του Huntington, οι παθομορφολογικές αλλαγές στο ραβδωτό σώμα είναι πιο έντονες και πιο εκτεταμένες, εμπλέκοντας τον εγκεφαλικό φλοιό, την παρεγκεφαλίδα, τον θάλαμο και την ωχρή σφαίρα.
Νευροχημικές αλλαγές στη νόσο του Huntington
GABA. Νευροχημικές μελέτες του εγκεφάλου σε ασθενείς με νόσο του Huntington αποκάλυψαν σημαντική μείωση της συγκέντρωσης GABA στο ραβδωτό σώμα. Μεταγενέστερες μελέτες επιβεβαίωσαν ότι η νόσος του Huntington σχετίζεται με μείωση του αριθμού των GABAεργικών νευρώνων και έδειξαν ότι οι συγκεντρώσεις GABA μειώνονται όχι μόνο στο ραβδωτό σώμα αλλά και στις ζώνες προβολής του - τα εξωτερικά και εσωτερικά τμήματα της ωχράς σφαίρας και της μέλαινας ουσίας. Στον εγκέφαλο στη νόσο του Huntington, ανιχνεύθηκαν επίσης αλλαγές στους υποδοχείς GABA χρησιμοποιώντας μελέτες δέσμευσης υποδοχέων και υβριδισμό in situ του mRNA. Ο αριθμός των υποδοχέων GABA μειώθηκε μέτρια στον κερκοφόρο πυρήνα και στο κέλυφος, αλλά αυξήθηκε στο δικτυωτό τμήμα της μέλαινας ουσίας και στο εξωτερικό τμήμα της ωχράς σφαίρας, κάτι που πιθανώς οφείλεται σε υπερευαισθησία στην απονεύρωση.
Ακετυλοχολίνη. Η ακετυλοχολίνη χρησιμοποιείται ως νευροδιαβιβαστής από μεγάλους μη ακανθώδεις νευρώνες στο ραβδωτό σώμα. Πρώιμες μεταθανάτιες μελέτες σε ασθενείς με νόσο του Huntington έδειξαν μειωμένη δραστικότητα της ακετυλοτρανσφεράσης της χολίνης (ChAT) στο ραβδωτό σώμα, υποδηλώνοντας απώλεια χολινεργικών νευρώνων. Ωστόσο, σε σύγκριση με τη σημαντική μείωση των GABAεργικών νευρώνων, οι χολινεργικοί νευρώνες είναι σχετικά διασώζονται. Επομένως, η πυκνότητα των θετικών για ακετυλοχολινεστεράση νευρώνων και η δραστικότητα ChAT στο ραβδωτό σώμα είναι στην πραγματικότητα σχετικά αυξημένες σε σύγκριση με τους μάρτυρες αντίστοιχης ηλικίας.
Ουσία P. Η ουσία P περιέχεται σε πολλούς μεσαίου μεγέθους ακανθώδεις νευρώνες του ραβδωτού σώματος, οι οποίοι προεξέχουν κυρίως προς το εσωτερικό τμήμα της ωχράς σφαίρας και της μέλαινας ουσίας και συνήθως περιέχουν επίσης δυνορφίνη και GABA. Τα επίπεδα της ουσίας P στο ραβδωτό σώμα και στον δικτυωτό πόρο της μέλαινας ουσίας μειώνονται στη νόσο του Huntington. Στο τελικό στάδιο της νόσου, ανοσοϊστοχημικές μελέτες έχουν αποκαλύψει σημαντική μείωση στον αριθμό των νευρώνων που περιέχουν την ουσία P. Σε προηγούμενα στάδια, οι νευρώνες που περιέχουν την ουσία P και προεξέχουν προς το εσωτερικό τμήμα της ωχράς σφαίρας είναι σχετικά απαλλαγμένοι, σε σύγκριση με τους νευρώνες που προεξέχουν προς τον δικτυωτό πόρο της μέλαινας ουσίας.
Οπιοειδή πεπτίδια. Η εγκεφαλίνη περιέχεται στους GABAεργικούς νευρώνες μεσαίας ακανθώδους προβολής της έμμεσης οδού, οι οποίοι προεξέχουν στο εξωτερικό τμήμα της ωχράς σφαίρας και φέρουν υποδοχείς D2. Ανοσοϊστοχημικές μελέτες έχουν δείξει ότι οι νευρώνες που περιέχουν εγκεφαλίνη και προεξέχουν στο εξωτερικό τμήμα της ωχράς σφαίρας χάνονται νωρίς στη νόσο του Huntington. Αυτά τα κύτταρα προφανώς πεθαίνουν νωρίτερα από τα κύτταρα που περιέχουν ουσία P και προεξέχουν στο εσωτερικό τμήμα της ωχράς σφαίρας.
Κατεχολαμίνες. Οι νευρώνες που περιέχουν βιογενείς αμίνες (ντοπαμίνη, σεροτονίνη) και προεξέχουν προς το ραβδωτό σώμα βρίσκονται στο συμπαγές τμήμα της μέλαινας ουσίας, στο κοιλιακό καλύπτρα και στους πυρήνες της ραφής. Ενώ οι νοραδρενεργικές προβολές προς το ανθρώπινο ραβδωτό σώμα είναι ελάχιστες, τα επίπεδα σεροτονίνης και ντοπαμίνης (ανά γραμμάριο ιστού) στο ραβδωτό σώμα είναι αυξημένα, υποδεικνύοντας τη διατήρηση αυτών των προσαγωγών προβολών παρά την αξιοσημείωτη απώλεια των νευρώνων του ίδιου του ραβδωτού σώματος. Οι ντοπαμινεργικοί νευρώνες της μέλαινας ουσίας παραμένουν άθικτοι τόσο στις κλασικές όσο και στις νεανικές μορφές της νόσου του Huntington.
Σωματοστατίνη/νευροπεπτίδιο Υ και συνθετάση νιτρικού οξειδίου. Η μέτρηση των επιπέδων σωματοστατίνης και νευροπεπτιδίου Υ στο ραβδωτό σώμα στη νόσο του Huntington αποκάλυψε 4-5 φορές αύξηση σε σύγκριση με τους φυσιολογικούς ιστούς. Ανοσοϊστοχημικές μελέτες έδειξαν απόλυτη διατήρηση των νευρώνων του ραβδωτού σώματος που περιέχουν νευροπεπτίδιο Υ, σωματοστατίνη και συνθετάση νιτρικού οξειδίου. Έτσι, αυτοί οι νευρώνες είναι ανθεκτικοί στην παθολογική διαδικασία.
Διεγερτικά αμινοξέα. Έχει προταθεί ότι ο επιλεκτικός κυτταρικός θάνατος στη νόσο του Huntington οφείλεται σε νευροτοξική δράση που προκαλείται από το γλουταμινικό. Τα επίπεδα γλουταμινικού και κινολινικού οξέος (μια ενδογενής νευροτοξίνη που είναι υποπροϊόν του μεταβολισμού της σεροτονίνης και αγωνιστής των υποδοχέων γλουταμινικού) στο ραβδωτό σώμα της νόσου του Huntington είναι ελαφρώς μεταβαλλόμενα, αλλά μια πρόσφατη μελέτη που χρησιμοποίησε φασματοσκοπία μαγνητικής τομογραφίας αποκάλυψε αύξηση στα επίπεδα γλουταμινικού in vivo. Το επίπεδο του νευρογλοιακού ενζύμου που είναι υπεύθυνο για τη σύνθεση του κινολινικού οξέος στο ραβδωτό σώμα της νόσου του Huntington αυξάνεται περίπου 5 φορές σε σύγκριση με το φυσιολογικό, ενώ η δραστικότητα του ενζύμου που εξασφαλίζει την αποικοδόμηση του κινολινικού οξέος αυξάνεται στη νόσο του Huntington μόνο κατά 20-50%. Έτσι, η σύνθεση του κινολινικού οξέος μπορεί να αυξηθεί στη νόσο του Huntington.
Μελέτες υποδοχέων διεγερτικών αμινοξέων (EAA) στη νόσο του Huntington έχουν αποκαλύψει σημαντική μείωση στον αριθμό των υποδοχέων NMDA, AMPA, καϊνικού και μεταβοτρόπου γλουταμινικού στο ραβδωτό σώμα, καθώς και των υποδοχέων AMPA και καϊνικού στον εγκεφαλικό φλοιό. Στο τελικό στάδιο της νόσου του Huntington, οι υποδοχείς NMDA ήταν ουσιαστικά απόντες, ενώ στα προκλινικά και πρώιμα στάδια, παρατηρήθηκε σημαντική μείωση στον αριθμό αυτών των υποδοχέων.
Επιλεκτική ευαισθησία. Στη νόσο του Huntington, ορισμένοι τύποι ραβδωτών κυττάρων χάνονται επιλεκτικά. Οι μεσαίου μεγέθους ακανθώδεις νευρώνες, οι οποίοι προεξέχουν στο εξωτερικό τμήμα της ωχράς σφαίρας και περιέχουν GABA και εγκεφαλίνη, πεθαίνουν πολύ νωρίς στη νόσο, όπως και οι νευρώνες που περιέχουν GABA και ουσία P και προεξέχουν στο δικτυωτό τμήμα της μέλαινας ουσίας. Η απώλεια νευρώνων που περιέχουν GABA και εγκεφαλίνη και προεξέχουν στο εξωτερικό τμήμα της ωχράς σφαίρας αποαναστέλλει αυτή τη δομή, η οποία με τη σειρά της οδηγεί σε ενεργή αναστολή του υποθαλαμικού πυρήνα. Η μειωμένη δραστηριότητα του υποθαλαμικού πυρήνα μπορεί προφανώς να εξηγήσει τις χορειόμορφες κινήσεις που εμφανίζονται στη νόσο του Huntington. Είναι γνωστό εδώ και καιρό ότι οι εστιακές αλλοιώσεις του υποθαλαμικού πυρήνα μπορούν να προκαλέσουν χορεία. Η απώλεια νευρώνων GABA και ουσίας P που προεξέχουν στο δικτυωτό τμήμα της μέλαινας ουσίας είναι πιθανό να ευθύνεται για τις οφθαλμοκινητικές διαταραχές που παρατηρούνται στη νόσο του Huntington. Αυτή η οδός κανονικά αναστέλλει τους νευρώνες της μέλαινας ουσίας του δικτυωτού τμήματος που προεξέχουν προς το άνω διδύμιο, οι οποίοι με τη σειρά τους ρυθμίζουν τις σακκαδικές κινήσεις. Στη νεανική νόσο του Huntington, οι προαναφερθείσες οδοί επηρεάζονται σοβαρότερα και, επιπλέον, οι ραβδωτές προβολές προς το εσωτερικό τμήμα της ωχράς σφαίρας χάνονται νωρίς.
Η πρωτεΐνη huntingtin, η οποία κωδικοποιείται από το γονίδιο του οποίου η μετάλλαξη προκαλεί τη νόσο του Huntington, βρίσκεται σε διάφορες δομές του εγκεφάλου και άλλων ιστών. Η huntingtin βρίσκεται συνήθως κυρίως στο κυτταρόπλασμα των νευρώνων. Η πρωτεΐνη βρίσκεται στους περισσότερους νευρώνες του εγκεφάλου, αλλά πρόσφατα δεδομένα δείχνουν ότι η περιεκτικότητά της είναι υψηλότερη στους νευρώνες της μήτρας από ό,τι στους νευρώνες των striosomal και υψηλότερη στους νευρώνες προβολής από ό,τι στους μεσονευρώνες. Έτσι, η επιλεκτική ευαισθησία των νευρώνων συσχετίζεται με την περιεκτικότητά τους σε huntingtin, η οποία υπάρχει κανονικά σε ορισμένους νευρωνικούς πληθυσμούς.
Όπως και στους εγκεφάλους ασθενών με νόσο του Huntington, σε ποντίκια που είναι διαγονιδιακά για το Ν-τελικό θραύσμα του γονιδίου της νόσου του Huntington με αυξημένο αριθμό επαναλήψεων, η huntingtin σχηματίζει πυκνά συσσωματώματα στους πυρήνες των νευρώνων. Αυτά τα ενδοπυρηνικά εγκλείσματα σχηματίζονται σε νευρώνες προβολής ραβδωτού σώματος (αλλά όχι σε μεσονευρώνες). Σε διαγονιδιακά ποντίκια, τα εγκλείσματα σχηματίζονται αρκετές εβδομάδες πριν από την έναρξη των συμπτωμάτων. Αυτά τα δεδομένα υποδηλώνουν ότι η πρωτεΐνη huntingtin που περιέχει αυξημένο αριθμό υπολειμμάτων γλουταμίνης των οποίων τα εγκλείσματα κωδικοποιούν επαναλήψεις τρινουκλεοτιδίων ή ένα θραύσμα αυτών, συσσωρεύεται στον πυρήνα και κατά συνέπεια μπορεί να επηρεάσει τον έλεγχο των κυτταρικών λειτουργιών.
Συμπτώματα της νόσου του Χάντινγκτον
Η ηλικία στην οποία εμφανίστηκαν τα πρώτα συμπτώματα σε ασθενείς με νόσο του Huntington είναι δύσκολο να προσδιοριστεί με ακρίβεια, καθώς η νόσος εκδηλώνεται σταδιακά. Αλλαγές στην προσωπικότητα και τη συμπεριφορά, ήπιες διαταραχές συντονισμού μπορεί να εμφανιστούν πολλά χρόνια πριν από την εμφάνιση πιο εμφανών συμπτωμάτων. Μέχρι να τεθεί η διάγνωση, οι περισσότεροι ασθενείς έχουν χορειακές κινήσεις, μειωμένο συντονισμό λεπτών κινήσεων και αργή παραγωγή εκούσιων σακκαδικών κινήσεων. Καθώς η νόσος εξελίσσεται, η ικανότητα οργάνωσης των δραστηριοτήτων μειώνεται, η μνήμη μειώνεται, η ομιλία γίνεται δύσκολη, οι οφθαλμοκινητικές διαταραχές και η μειωμένη απόδοση συντονισμένων κινήσεων αυξάνονται. Αν και στο πρώιμο στάδιο της νόσου δεν υπάρχουν αλλαγές στους μύες και τη στάση του σώματος, καθώς εξελίσσεται, μπορεί να αναπτυχθούν δυστονικές στάσεις, οι οποίες με την πάροδο του χρόνου μπορεί να μετατραπούν σε κυρίαρχο σύμπτωμα. Σε μεταγενέστερο στάδιο, η ομιλία γίνεται δυσλειτουργική, η κατάποση γίνεται σημαντικά δύσκολη, το περπάτημα γίνεται αδύνατο. Η νόσος του Huntington συνήθως εξελίσσεται σε διάστημα 15-20 ετών. Στο τελικό στάδιο, ο ασθενής είναι αβοήθητος και χρειάζεται συνεχή φροντίδα. Η μοιραία έκβαση δεν σχετίζεται άμεσα με την πρωτοπαθή νόσο, αλλά με τις επιπλοκές της, για παράδειγμα, την πνευμονία.
Άνοια στη νόσο του Huntington
Κωδικός ICD-10
P02.2. Άνοια στη νόσο του Huntington (G10).
Η άνοια αναπτύσσεται ως μία από τις εκδηλώσεις μιας συστηματικής εκφυλιστικής-ατροφικής διαδικασίας με κυρίαρχη βλάβη στο ραβδωτό σύστημα του εγκεφάλου και σε άλλους υποκοιλιακούς πυρήνες. Κληρονομείται με αυτοσωμικό επικρατή τρόπο.
Κατά κανόνα, η νόσος εκδηλώνεται στην τρίτη ή τέταρτη δεκαετία της ζωής με χορεόμορφη υπερκινητικότητα (ειδικά στο πρόσωπο, τα χέρια, τους ώμους, το βάδισμα), αλλαγές στην προσωπικότητα (διεγερτικοί, υστερικοί και σχιζοειδείς τύποι ανωμαλιών προσωπικότητας), ψυχωσικές διαταραχές (ειδική κατάθλιψη με κατήφεια, σκυθρωπότητα, δυσφορία, παρανοϊκή διάθεση).
Ιδιαίτερη σημασία για τη διάγνωση έχει ο συνδυασμός χορεόμορφης υπερκινητικότητας, άνοιας και κληρονομικού φορτίου. Τα ακόλουθα είναι ειδικά για αυτήν την άνοια:
- αργή εξέλιξη (κατά μέσο όρο 10-15 χρόνια): αποσύνδεση μεταξύ της εναπομένουσας ικανότητας αυτοφροντίδας και της προφανούς πνευματικής ανικανότητας σε καταστάσεις που απαιτούν παραγωγική πνευματική εργασία (εννοιολογική σκέψη, εκμάθηση νέων πραγμάτων)·
- έντονη ανομοιομορφία της ψυχικής απόδοσης, η οποία βασίζεται σε σοβαρές διαταραχές της προσοχής και αστάθεια στις στάσεις του ασθενούς ("σπασμωδική" σκέψη, παρόμοια με την υπερκινητικότητα).
- ατυπία εμφανών παραβιάσεων των ανώτερων φλοιωδών λειτουργιών.
- αντίστροφη σχέση μεταξύ της αύξησης της άνοιας και της σοβαρότητας των ψυχωσικών διαταραχών.
Λαμβάνοντας υπόψη το υψηλό ποσοστό ψυχωτικών (παρανοϊκές παραληρητικές ιδέες ζήλιας, καταδίωξης) και δυσφορικών διαταραχών στην κλινική εικόνα της νόσου, η θεραπεία πραγματοποιείται χρησιμοποιώντας διάφορα νευροληπτικά που μπλοκάρουν τους ντοπαμινεργικούς υποδοχείς (παράγωγα φαινοθειαζίνης και βουτυροφαινόνης) ή μειώνουν το επίπεδο ντοπαμίνης στους ιστούς (ρεσερπίνη).
Χρησιμοποιούνται αλοπεριδόλη (2-20 mg/ημέρα), τιαπρίδη (100-600 mg/ημέρα) για όχι περισσότερο από τρεις μήνες, θειοριδαζίνη (έως 100 mg/ημέρα), ρεσερπίνη (0,25-2 mg/ημέρα) και το αντισπασμωδικό κλοναζεπάμη (1,5-6 mg/ημέρα). Αυτά τα φάρμακα βοηθούν στη μείωση της υπερκινητικότητας, στην εξομάλυνση της συναισθηματικής έντασης και στην αντιστάθμιση των διαταραχών προσωπικότητας.
Η ενδονοσοκομειακή θεραπεία των ψυχικών διαταραχών πραγματοποιείται λαμβάνοντας υπόψη το κύριο σύνδρομο, την ηλικία και τη γενική κατάσταση του ασθενούς. Στην εξωτερική θεραπεία, οι αρχές της θεραπείας είναι οι ίδιες (συνεχής θεραπεία συντήρησης των κινητικών διαταραχών, περιοδική αλλαγή φαρμάκου). Στην εξωτερική θεραπεία χρησιμοποιούνται χαμηλότερες δόσεις νευροληπτικών.
Τα μέτρα αποκατάστασης για την ήπια και μέτρια άνοια περιλαμβάνουν εργοθεραπεία, ψυχοθεραπεία και γνωστική εκπαίδευση. Είναι απαραίτητο να συνεργαστείτε με τα μέλη της οικογένειας και να παρέχετε ψυχολογική υποστήριξη στα άτομα που φροντίζουν τον ασθενή. Η κύρια μέθοδος πρόληψης ασθενειών είναι η ιατρική και γενετική συμβουλευτική των στενότερων συγγενών του ασθενούς με παραπομπή για ανάλυση DNA κατά τη λήψη απόφασης για την τεκνοποίηση.
Η πρόγνωση είναι γενικά δυσμενής. Η πορεία της νόσου είναι αργά εξελισσόμενη και συνήθως οδηγεί σε θάνατο μετά από 10-15 χρόνια.
[ 18 ]
Τι σε προβληματιζει?
Διάγνωση της νόσου του Huntington
Η διάγνωση βασίζεται σε τυπικά συμπτώματα, οικογενειακό ιστορικό και γενετικό έλεγχο. Λόγω ατροφίας της κεφαλής του κερκοφόρου πυρήνα, η μαγνητική τομογραφία και η καρδιαγγειακή τομογραφία αποκαλύπτουν διόγκωση των εγκεφαλικών κοιλιών στο τελικό στάδιο της νόσου.
Θεραπεία της νόσου του Huntington
Η θεραπεία της νόσου του Huntington είναι συμπτωματική. Η χορεία και η διέγερση μπορούν να κατασταλούν μερικώς με νευροληπτικά (π.χ. χλωροπρομαζίνη 25-300 mg από το στόμα 3 φορές την ημέρα, αλοπεριδόλη 5-45 mg από το στόμα 2 φορές την ημέρα) ή ρεσερπίνη 0,1 mg από το στόμα μία φορά την ημέρα. Οι δόσεις αυξάνονται στο μέγιστο ανεκτό (πριν εμφανιστούν παρενέργειες, όπως υπνηλία, παρκινσονισμός· για τη ρεσερπίνη, υπόταση). Ο στόχος της εμπειρικής θεραπείας είναι η μείωση της γλουταμινεργικής διαβίβασης μέσω υποδοχέων N-μεθυλο-Ο-ασπαρτικού και η διατήρηση της παραγωγής ενέργειας στα μιτοχόνδρια. Η θεραπεία που στοχεύει στην αύξηση του GABA στον εγκέφαλο είναι αναποτελεσματική.
Ο γενετικός έλεγχος και η συμβουλευτική είναι σημαντικά, επειδή τα συμπτώματα της νόσου εμφανίζονται μετά την αναπαραγωγική ηλικία. Άτομα με θετικό οικογενειακό ιστορικό και όσοι ενδιαφέρονται για τον έλεγχο παραπέμπονται σε εξειδικευμένα κέντρα, λαμβάνοντας υπόψη όλες τις ηθικές και ψυχολογικές επιπτώσεις.
Συμπτωματική θεραπεία της νόσου του Huntington
Δεν υπάρχει αποτελεσματική θεραπεία που να μπορεί να σταματήσει την εξέλιξη της νόσου του Huntington. Έχουν διεξαχθεί αρκετές δοκιμές διαφόρων φαρμάκων, αλλά δεν έχει επιτευχθεί σημαντικό αποτέλεσμα. Τα νευροληπτικά και άλλοι ανταγωνιστές υποδοχέων ντοπαμίνης χρησιμοποιούνται ευρέως για τη διόρθωση ψυχικών διαταραχών και ακούσιων κινήσεων σε ασθενείς με νόσο του Huntington. Οι ακούσιες κινήσεις αντανακλούν μια ανισορροπία μεταξύ του ντοπαμινεργικού και του GABAεργικού συστήματος. Συνεπώς, τα νευροληπτικά χρησιμοποιούνται για τη μείωση της υπερβολικής ντοπαμινεργικής δραστηριότητας. Ωστόσο, αυτά τα φάρμακα από μόνα τους μπορούν να προκαλέσουν σημαντικές γνωστικές και εξωπυραμιδικές παρενέργειες. Επιπλέον, εκτός από τις περιπτώσεις όπου ο ασθενής εμφανίζει ψύχωση ή διέγερση, η αποτελεσματικότητά τους δεν έχει αποδειχθεί. Τα νευροληπτικά συχνά προκαλούν ή επιδεινώνουν τη δυσφαγία ή άλλες κινητικές διαταραχές. Τα νευροληπτικά νεότερης γενιάς όπως η ρισπεριδόνη, η κλοζαπίνη και η ολανζαπίνη μπορεί να είναι ιδιαίτερα χρήσιμα στη θεραπεία της νόσου του Huntington επειδή προκαλούν λιγότερες εξωπυραμιδικές παρενέργειες, αλλά μπορεί να μειώσουν τα παρανοϊκά συμπτώματα ή την αυξημένη ευερεθιστότητα.
Η τετραβεναζίνη και η ρεσερπίνη μειώνουν επίσης τη δραστηριότητα του ντοπαμινεργικού συστήματος και μπορούν να μειώσουν τη σοβαρότητα των ακούσιων κινήσεων στα πρώιμα στάδια της νόσου. Ωστόσο, αυτά τα φάρμακα μπορούν να προκαλέσουν κατάθλιψη. Δεδομένου ότι η ίδια η νόσος συχνά προκαλεί κατάθλιψη, αυτή η παρενέργεια περιορίζει σημαντικά τη χρήση της ρεσερπίνης και της τετραβεναζίνης. Στα τελευταία στάδια της νόσου, τα κύτταρα που φέρουν υποδοχείς ντοπαμίνης πεθαίνουν, επομένως η αποτελεσματικότητα των ανταγωνιστών των υποδοχέων ντοπαμίνης εξασθενεί ή χάνεται.
Τα νευροληπτικά, τα αντικαταθλιπτικά και τα αγχολυτικά χρησιμοποιούνται για τη θεραπεία της ψύχωσης, της κατάθλιψης και της ευερεθιστότητας σε ασθενείς με νόσο του Huntington, αλλά θα πρέπει να συνταγογραφούνται μόνο για όσο διάστημα ο ασθενής έχει πραγματικά αυτά τα συμπτώματα. Τα φάρμακα που μπορεί να είναι χρήσιμα σε ένα στάδιο της νόσου μπορεί να καταστούν αναποτελεσματικά ή ακόμη και επιβλαβή καθώς η νόσος εξελίσσεται.
Οι αγωνιστές των υποδοχέων GABA έχουν δοκιμαστεί σε ασθενείς με νόσο του Huntington, καθώς η νόσος του Huntington έχει αποδειχθεί ότι παρουσιάζει σημαντική μείωση των επιπέδων GABA στο ραβδωτό σώμα, καθώς και υπερευαισθησία των υποδοχέων GABA στις περιοχές προβολής του. Οι βενζοδιαζεπίνες έχουν αποδειχθεί αποτελεσματικές σε περιπτώσεις όπου οι ακούσιες κινήσεις και η γνωστική εξασθένηση επιδεινώνονται από το στρες και το άγχος. Θα πρέπει να συνταγογραφούνται χαμηλές δόσεις αυτών των φαρμάκων για την αποφυγή ανεπιθύμητων ηρεμιστικών επιδράσεων. Στους περισσότερους ασθενείς με νόσο του Huntington, κανένα από τα φάρμακα δεν οδηγεί σε σημαντική βελτίωση της ποιότητας ζωής.
Στην πρώιμη έναρξη της νόσου του Huntington με παρκινσονικά συμπτώματα, μπορούν να δοκιμαστούν ντοπαμινεργικοί παράγοντες, αλλά η αποτελεσματικότητά τους είναι περιορισμένη. Επιπλέον, η λεβοντόπα μπορεί να προκαλέσει ή να αυξήσει τον μυόκλονο σε αυτούς τους ασθενείς. Ταυτόχρονα, η βακλοφαίνη μπορεί να μειώσει την ακαμψία σε ορισμένους ασθενείς με νόσο του Huntington.
[ 26 ], [ 27 ], [ 28 ], [ 29 ]
Προληπτική (νευροπροστατευτική) θεραπεία της νόσου του Huntington
Παρόλο που το γενετικό ελάττωμα στη νόσο του Huntington είναι γνωστό, ο τρόπος με τον οποίο οδηγεί σε επιλεκτική νευρωνική εκφύλιση παραμένει ασαφής. Υποτίθεται ότι οι προληπτικές θεραπείες που στοχεύουν στη μείωση του οξειδωτικού στρες και της διεγερτικής τοξικότητας ενδέχεται να επιβραδύνουν ή να σταματήσουν την εξέλιξη της νόσου. Η κατάσταση μπορεί να είναι κάπως παρόμοια με την ηπατοφακοειδή εκφύλιση, στην οποία το γενετικό ελάττωμα παρέμεινε άγνωστο για πολλά χρόνια, αλλά οι προληπτικές θεραπείες που στοχεύουν στο δευτερογενές αποτέλεσμα, τη συσσώρευση χαλκού, «θεραπεύτηκαν». Από αυτή την άποψη, η υπόθεση ότι η νόσος του Huntington σχετίζεται με μια διαταραχή του ενεργειακού μεταβολισμού και τον κυτταρικό θάνατο λόγω διεγερτικής τοξικότητας έχει προσελκύσει ιδιαίτερη προσοχή. Η ίδια η νόσος μπορεί να προκαλέσει κυτταρικό θάνατο λόγω ενδοπυρηνικής συσσωμάτωσης Ν-τελικών θραυσμάτων huntingtin, η οποία διαταράσσει τις κυτταρικές και μεταβολικές λειτουργίες. Αυτή η διαδικασία μπορεί να επηρεάσει ορισμένες ομάδες νευρώνων σε μεγαλύτερο βαθμό από άλλες λόγω της υψηλότερης ευαισθησίας τους στη διεγερτική τοξική βλάβη. Σε αυτήν την περίπτωση, η προληπτική θεραπεία με ανταγωνιστές υποδοχέων διεγερτικών αμινοξέων ή παράγοντες που αποτρέπουν τη βλάβη από τις ελεύθερες ρίζες θα είναι σε θέση να αποτρέψει ή να καθυστερήσει την έναρξη και την εξέλιξη της νόσου. Σε εργαστηριακά μοντέλα αμυοτροφικής πλευρικής σκλήρυνσης, έχει αποδειχθεί ότι οι αντιοξειδωτικοί παράγοντες και οι ανταγωνιστές υποδοχέων (RAA) είναι σε θέση να επιβραδύνουν την εξέλιξη της νόσου. Παρόμοιες προσεγγίσεις μπορεί να είναι αποτελεσματικές στη νόσο του Huntington. Κλινικές δοκιμές ανταγωνιστών υποδοχέων γλουταμινικού και παραγόντων που ενισχύουν τη λειτουργία του συμπλόκου II της μιτοχονδριακής αλυσίδας μεταφοράς ηλεκτρονίων βρίσκονται σε εξέλιξη.
[ 30 ], [ 31 ], [ 32 ], [ 33 ], [ 34 ], [ 35 ], [ 36 ], [ 37 ], [ 38 ], [ 39 ]